Research Article
Whole transcriptome cerebral cortex gene analysis in Alzheimer’s diseases 
2 Division of Bioinformatics, Scientific Bio-Minds, Bangalore, India
 Author
Author  Correspondence author
Correspondence author
Computational Molecular Biology, 2016, Vol. 6, No. 2 doi: 10.5376/cmb.2016.06.0002
Received: 25 Dec., 2015 Accepted: 21 Mar., 2016 Published: 29 Mar., 2016
Pooja S., Muttagi A.P., Patel S.J., Gurumurthy H., and Prashantha C.N., 2016, Whole transcriptome cerebral cortex gene analysis in Alzheimer’s diseases, Computational Molecular Biology, 6(2): 1-9
Neurodegenerative disorder such as Alzheimer's is most common form of memory loss observed on aging population. The genetic factors are strongly influenced by risk of accumulation of β-amyloid protein, apolipoprotein (APOE4), intracellular neurofibrillary tangles of hyperphosphorylated tau proteins that form plaques in the extracellular region of brain. There is no clear method to diagnosis Alzheimer’s disease in the earliest stages, but there are few treatments available to reduce the symptoms but there is no clear cure for the disease. A Genetic markers, inflammatory markers and blood based protein markers that could predict onset on disease prognosis. There is a need to identify markers from the entire genome that influenced by interlink with gene-gene, gene-protein and gene-environment interactions. Different transcriptional factors such as amyloid precursor protein gene (Aβ) and the presenilin 1 and 2 genes are major risk for Alzheimer’s disease. Using computational techniques to identify susceptible genetic factors from functional genomics and proteomics to understand transcriptional coregulation and transcription factor binding sites which potentially contribute to Alzheimer’s disease.
1 Introduction
Alzheimer’s disease (AD) is a neurological disorder causing dementia and memory loss in the Aging people (Breteler, 1992). The protocatalytic characteristic of beta amyloid precursor protein (APP) and apolipoprotein ApoE has familial mutations that associated regulation of familial and sporadic AD (Christensen et al., 2004). The research evidences that helps to predict 40-42 amino-acid peptides generates multiple proteolytic cleavage of the APP that triggers numerous pathophysiological changes that toxic oligomers of APP, the overall function is remain unknown (Zheng and Koo, 2011). The genetic variants encoding an alanine-to-threonine substitution at residue 673 (A673T) that confers mutations in AD (Jonsson et al., 2012). The mutations in presenilin1 (PSEN1) and presenilin 2 (PSEN2) enzymes involved in transcriptional regulation to synthesize protein that may processing of APP to develop familial AD (Cruts et al., 1998; Hardy et al., 2001; Kauwe et al., 2007). More recently, PSEN1 mutations have also been identified in some sporadic cases of late onset AD (Cruchaga et al., 2012). There are so many number of gene mutations in AD itself have also been identified many of these mutation clusters at or near the β or γ proteolytic sites, favoring either the overproduction of total amyloid-β (Aβ) (Citron et al., 1992; Di Fede et al., 2009, Zhou et al., 2011) or an increased ratio of the pro-aggregating Aβ1-42 species relative to Aβ1-40 (De Jonghe et al., 2001; Kwok et al., 2000; Scheuner et al., 1996; Suzuki et al., 1994). In other instances, mutations within the Aβ peptide promote an increased propensity for aggregation (Nilsberth et al., 2001; Tomiyama et al., 2008). Together, these genetic findings provide strong support for the amyloid hypothesis of AD, which postulates that an imbalance in the production and clearance of Aβ initiates a cascade of amyloid accumulation, neurotoxicity and neurodegeneration (Ertekin-Taner, 2008). Although it is clear that expression levels of AD genes are important in AD etiology, much remains unknown about their specific regulation (Theuns and Van Broeckhoven, 2000).
The Apolipoprotein E (ApoE) is a major cholesterol carrier in the blood-brain barrier (Liu et al., 2013; Herz and Chen, 2006). ApoE is primarily produced by astrocytes and its function is to deliver lipids to neurons through the binding of cell surface ApoE receptors (Chen et al., 2011). Human ApoE exists as three polymorphic alleles: ε2, ε3 and ε4 (Frieden and Garai, 2012). These three allele forms differ from each other in single amino acid change, resulting in different protein structures, lipid association and receptor binding (Zhong and Weisgraber, 2006; Bu, 2006; Corder et al., 1993). The ε4 allele of the ApoE is the strongest genetic risk factor for late-onset AD (LOAD) (Farrer et al., 1997). Individuals with one ε4 allele are 3–4 times more likely to develop AD than those without ε4 allele (Laws et al., 2003). Interestingly, the rare ε2 allele has a protective effect against AD compared with the ε3 allele (Taddei et al., 1997). Studying the regulatory elements of disease genes and their corresponding transcription factors is therefore critically important for elucidation of the disease processes (Mahley, 1988). This review will discuss the mechanisms of transcriptional regulation for AD genes, and the misregulation that leads to AD susceptibility. A large number of genes that associated with disease, although there is a need to identify expression levels of genes, etiology and gene regulations and much functions remains unknown.
2 Materials and Methods
2.1 Data selection
The whole genome analysis of Alzheimer’s disease (AD) to identify the copy number variation and gene mutations. The histopathology of disease shows extracellular amyloid plaques and intracellular neurofibrillary tangles. The histopathology and pathophysiology of disease helps to identify the disease prognosis. There is a search for biomarkers of Alzheimer’s disease (AD) have yielded numerous expensive and/or invasive candidates, including putative disease markers. To assess the state of blood-based biomarkers, genetic markers and inflammatory markers for AD including the understanding the context of use to begin outlining the research challenges related to the development of markers including understanding the context of use from a clinical, research, and regulatory perspective; highlighting the need for standardization and harmonization of protocols; and identifying knowledge gaps and the research efforts needed to fill those gaps.
2.2 Microarray analysis
The systematic identification and characterization of Alzheimer’s disease (AD) specific transcriptome cerebral cortex gene analysis using microarray gene expression data, the dataset contains 31 samples of which 9 control and 22 AD affected samples obtained from Gene Expression Omnibus (GEO Accession Number: GDS1297) was used in this study. Gene expression was measured using GPL96 annotation file to compare the covering 22283 genes for AD (22 samples) and control (9 samples). The gene expression of control and AD stage has been considered and compared for the identification of differentially expressed genes in AD stage. Significance analysis of Microarray (SAM) determines the significant changes in expression of genes between different biological stages based on statistical analysis of modified gene specific t-test. We used MultiExperiment Viewer (MEV) software package from TIGR for hierarchical clustering of microarray data, using Euclidean Distance metrics and Average Linkage Clustering algorithms. The Tree View shows the relationship between the genes based on the gene expression profile.
2.3 Analysis of gene enrichment for transcriptome cerebral cortex genes
Each clusters of gene set was analyzed for enrichment of transcriptome cerebral cortex genes using the DAVID Database. The conserved non-coding regions of the promoters were searched for matches to all cerebral cortex profiles in the GOrilla database.
3 Results
The identification of blood, inflammatory and genetic markers involved in diagnosis of Alzheimer's disease in early state using transcriptome specific cerebral cortex brain tissue to identify the differential expressed genes, that acts as a potential markers. The selected dataset contains 22283 genes of 31 samples of which 9 controls and 22 disease samples. All the samples are classified into four groups based on pathogenesis, such as control 9, incipient 7, moderate 8, and severe 7. The selected samples are tested with two different conditions based on diagnostic tests such as MiniMental Status Examination (MMSE) and neurofibrillary tangle (NFT) scores across all 31 subjects regardless of diagnosis (Table 1). The quality control of dataset is the major concept of design and pre-process raw data that helps to identify differential gene expression. Using different mean and median calculations of BioConductor algorithms to predict pre-process the data to evaluate quality values that helps to normalize the data. RMA and MAS5 algorithms help to predict the pre-processing and quality prediction. These tests help to identify differentially expressed genes present in transcriptional response that significantly correlated with AD markers.
.png) Table 1 Alzheimer's disease samples used for analysis of differential gene expression analysis |
3.1 Differential gene expression analysis
Using Biostatistical package such as LIMMA is used to determine the differential gene expressions based on significance of p-values. We have identified 9921 genes is significantly associated with AD. While multiple comparisons of control, moderate, incipient and severe datasets is classified based on MMSE and NFT test scores. The significance analysis of microarray shows 5727 genes is significantly associated with GO terms. The fundamental gene expression data of identified genes whose patterns of expression differ according to phenotype and genotype of experimental conditions. The transcriptional control of class discriminate genes that differentially expressed in 4 groups such as control, Incipient, moderate and severe levels. There are different statistical calculations such as t-test of probable values of gene-by-gene expression in each array is analyzed with FDR values. We calculate the FDR of the false positives of expected to multiple comparisons divided by the false positive results found with worst case probability of genes identified (P<0.05) by correlation is significant because of the error from multiple testing (Table 2). The two ways ANOVA of significant calculation shows 1361 genes is either up-regulated or down regulated by more than 2 fold change between the 4 groups. ANOVA calculation of one or two groups is tested with different groups is tested based on linear models that are frequently used for assessing differential gene expression. Due to lack of information on coregulation of genes that computed with genes separately. The overall results shows 12 genes CA11, PTN, TBC1D2B, FAR2, LHCGR, EHD1, KCNA5, GPR22, WDFY3 and ITGBL1 is transcriptional regulation of cerebral cortex in AD and is potential targets for biomarkers to identify AD for diagnostics (Table 3a; Table 3b; Table 3c; Table 3d). The LHCGR is the clinical phenotype of AD is the best potential biomarkers against AD diagnostics. We sorted the more significantly expressed in all four conditions that is predicted in bar graphs The overall results of significant genes shows PTN, TBC1D2B, FAR2, LHCGR, EHD1, KCNA5 and GPR22, is more potentially expressed in control-moderate, control-severe, incipient-severe and moderate and severe.
.png) Table 2 Differentially expressed genes that significantly expressed in AD |
 Table 3a Control-moderate of unregulated differential expressed 13 genes in AD |
 Table 3b Control-Severe of unregulated differential gene expression of 214 genes |
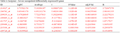 Table 3c Incipient - Severe unregulated differentially expressed 8 genes |
 Table 3d Moderate-Severe unregulated 65 genes |
3.2 Genome wide association studies
The combines studies of GWAS of control, incipient, moderate and severe samples are differently identified the gene expressions based on p-values. We have identified 12 genes that more significantly associated with transcriptional regulation of cerebral cortex and is potential targets against AD. Using GWAS studies of these genes which significantly involved in markers and is used for early stage detection of Alzheimer’s disease. The PTN gene is polymorphism associates with risk of age onset of AD. The PTN SNPs of rs4420638 within APOC1 was strongly linked with PTN of AD of p-value in the logistic of p<0.05. The other genes such as TBC1D2B, FAR2, LHCGR, EHD1, KCNA5, GPR22, WDFY3 and ITGBL1 genes also has significant SNPs that logistically associated with AD (Table 4).
 Table 4 GWAS analysis of disease genes predicted based on pharmacogenomic properties |
Using Pharmacogenomic properties of biomarkers prediction on based on gene-drug interaction. The PTN gene is interacting with Ozone co-treated with Chlorine results in increased expression of PTN mRNA. The TBC1D2B is strongly interacting with plant extracts results in increased expression of TBC1D2B mRNA to control AD. The LHCGR gene is strongly interacting with Melatonin, resveratrol and Sodium Fluoride has increase expression of mRNA to control the AD but the Particulate Matter are decreases the mRNA expression to regulate mRNA expression to stop the development of brain cells to control AD. The KCNA5 gene is interacting with Acrolein results in increased expression of KCNA5 gene that regulate the AD expression in transcriptional levels of cerebral cortex. Another drug such as Rotenon also interacts with KCNA5 that inhibit the reaction of transcription expression and stop ion gateway channels. Resveratrol affects the reaction geranylgeranylacetone results in increased expression of KCNA5 protein. Zinc deficiency results in decreased expression of KCNA5 mRNA. The WDFY3 gene is strongly interacting with APP protein modified form binds to Aluminum which results in decreased expression of WDFY3 mRNA. Nanotubes, Carbon co-treated with calfactant results in increased expression of WDFY3 protein. The overall results shows PTN, TBC1D1B, LHCGR, KCNA5 and WDFY5 are Pharmacologically interacting with different drugs that potentially targets for AD treatment.
4 Conclusions
Herein we discussed the differential gene expression analysis of Alzheimer’s disease that mainly expressed in transcriptional regulation of the cortex of the brain. There are 5527 genes that significantly associated with differential expression, these genes are further classified according to incipient, moderate, severe and normal tissues of expression levels shows only More severe condition level has the highest level of gene expression that significantly expressed in Alzheimer’s disease. Genome wide association of Alzheimer’s disease to predict the SNPs that transcriptionally associated with the cortex region of brain that regulate protein synthesis. We have identified 8 biomarkers such as PTN, TBC1D2B, FAR2, LHCGR, EHD1, KCNA5, WDFY3 and ITGBL1 is mainly involved transcriptional regulation and disease progression. Using Pharmacogenomic analysis of these genes has good effect for the treatment of AD and is potentially considered to regulate and also to modulate gene expression but also have important roles in diagnostics.
Acknowledgement
We gratefully acknowledge the assistance of Dr. C. N Prashantha for supporting project work, workflows, technical assistance and report preparation. We also grateful to Seema J Patel and Anitha P.Muttagi as internal guide to conduct seminars and guiding project. We also grateful to Department of Biotechnology, GMIT and Scientific Bio-Minds to providing continuous support to successfully complete the project.
Breteler M.M., Claus J.J., van Duijn C.M., Launer L.J., and Hofman A., 1992, Epidemiology of Alzheimer’s disease, Epidemiol. Rev., 14: 59–82.
Bu G., 2009, Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy, Nat Rev Neurosci, 10: 333–344.
http://dx.doi.org/10.1038/nrn2620
Christensen M.A., Zhou W., Qing H., Lehman A., Philipsen S., Song W., 2004, Transcriptional regulation of BACE1, the beta-amyloid precursor protein beta-secretase by Sp1, Mol Cell Biol, 24(2): 865-74
http://dx.doi.org/10.1128/MCB.24.2.865-874.2004
Cruts M., van Duijn C.M., Backhovens H., Van den Broeck M., Wehnert A., Serneels S., Sherrington R., Hutton M., Hardy J., St George-Hyslop P. H., Hofman A., and Van Broeckhoven C., 1998, Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease, Hum Mol Genet, 7: 43-51.
http://dx.doi.org/10.1093/hmg/7.1.43
Cruchaga C., Haller G., Chakraverty S., Mayo K., Vallania F.L., Mitra R.D., Faber K., Williamson J., Bird T., Diaz-Arrastia R., Foroud, T. M., Boeve B. F., Graff-Radford N.R., St Jean P., Lawson M., Ehm M.G., Mayeux R., and Goate A.M., 2012, Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer's disease families, PLoS One, 7: e31039.
http://dx.doi.org/10.1371/journal.pone.0031039
Citron M., Oltersdorf T., Haass C., McConlogue L., Hung A.Y., Seubert P., Vigo-Pelfrey C., Lieberburg I., and Selkoe D.J., 1992, Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production, Nature 360: 672-674.
http://dx.doi.org/10.1038/360672a0
Chen J., Li Q., and Wang J., 2011, Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions, Proc Natl Acad Sci USA, 108: 14813–14818.
http://dx.doi.org/10.1073/pnas.1106420108
Corder E.H., Saunders A.M., Strittmatter W.J., Schmechel D.E., Gaskell P.C., Small G.W., Roses A.D., Haines J.L., and Pericak-Vance M.A., 1993, Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families, Science, 261: 921–923.
http://dx.doi.org/10.1126/science.8346443
Di Fede G., Catania M., Morbin M., Rossi G., Suardi S., Mazzoleni G., Merlin M., Giovagnoli A.R., Prioni S., Erbetta A., Falcone C., Gobbi M., Colombo L., Bastone A., Beeg M., Manzoni C., Francescucci B., Spagnoli A., Cantu L., Del Favero E., Levy E., Salmona M., and Tagliavini, F., 2009, A recessive mutation in the APP gene with dominantnegative effect on amyloidogenesis, Science 323: 1473-1477.
http://dx.doi.org/10.1126/science.1168979
De Jonghe C., Esselens C., Kumar-Singh S., Craessaerts K., Serneels S., Checler F., Annaert W., Van Broeckhoven C., and De Strooper B., 2001, Pathogenic APP mutations near the gamma-secretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability, Hum Mol Genet 10: 1665-1671.
http://dx.doi.org/10.1093/hmg/10.16.1665
Ertekin-Taner N., 2011, Gene expression endophenotypes: a novel approach for gene discovery in Alzheimer's disease, Mol Neurodegener, 6: 31.
http://dx.doi.org/10.1186/1750-1326-6-31
Frieden C., and Garai K., 2012, Structural differences between apoE3 and apoE4 may be useful in developing therapeutic agents for Alzheimer's disease, Proc Natl Acad Sci USA, 109: 8913–8918.
http://dx.doi.org/10.1073/pnas.1207022109
Farrer L.A., Cupples L.A., Haines J.L., Hyman B., Kukull W.A., Mayeux R., Myers R.H., Pericak-Vance M.A., Risch N., and van Duijn C.M., 1997, Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease meta analysis consortium, JAMA, 278: 1349–1356.
http://dx.doi.org/10.1001/jama.1997.03550160069041
Hardy J., and Crook R., 2001, Presenilin mutations line up along transmembrane alpha-helices, Neurosci Lett, 306: 203-205.
http://dx.doi.org/10.1016/S0304-3940(01)01910-3
Herz J., and Chen Y., 2006, Reelin, lipoprotein receptors and synaptic plasticity, Nat Rev Neurosci, 7: 850–859.
http://dx.doi.org/10.1038/nrn2009
Jonsson T., Atwal J.K., Steinberg S., Snaedal J., Jonsson P.V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., Graham R.R., Huttenlocher J., Bjornsdottir G., Andreassen O.A., Jonsson E.G., Palotie A., Behrens T.W., Magnusson O.T., Kong A., Thorsteinsdottir U., Watts R.J., and Stefansson K., 2012, A mutation in APP protects against Alzheimer's disease and age-related cognitive decline, Nature 488: 96-99.
http://dx.doi.org/10.1038/nature11283
Kauwe J.S., Jacquart S., Chakraverty S., Wang J., Mayo K., Fagan A.M., Holtzman D.M., Morris J.C., and Goate A.M., 2007, Extreme cerebrospinal fluid amyloid beta levels identify family with late-onset Alzheimer's disease presenilin 1 mutation, Annals of neurology, 61: 446-453.
http://dx.doi.org/10.1002/ana.21099
Kwok J.B., Li Q.X., Hallupp M., Whyte S., Ames D., Beyreuther K., Masters C.L., and Schofield P.R., 2000, Novel Leu723Pro amyloid precursor protein mutation increases amyloid beta42(43) peptide levels and induces apoptosis, Annals of neurology, 47: 249-253.
http://dx.doi.org/10.1002/1531-8249(200002)47:2%3C249::AID-ANA18%3E3.0.CO;2-8
Liu C.C., Kanekiyo T., Xu H., and Bu G., 2013, Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy, Nat Rev Neurol, 9: 106–118.
http://dx.doi.org/10.1038/nrneurol.2012.263
Laws S.M., Hone E., Gandy S., and Martins R.N., 2003, Expanding the association between the APOE gene and the risk of Alzheimer's disease: possible roles for APOE promoter polymorphisms and alterations in APOE transcription, J Neurochem, 84: 1215–1236.
http://dx.doi.org/10.1046/j.1471-4159.2003.01615.x
Mahley R.W., 1988, Apolipoprotein E: cholesterol transport protein with expanding role in cell biology, Science, 240: 622–630.
http://dx.doi.org/10.1126/science.3283935
Nilsberth C., Westlind-Danielsson A., Eckman C.B., Condron M.M., Axelman K., Forsell C., Stenh C., Luthman J., Teplow D.B., Younkin S.G., Naslund J., and Lannfelt L., 2001, The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation, Nat Neurosci, 4: 887-893.
http://dx.doi.org/10.1038/nn0901-887
Scheuner D., Eckman C., Jensen M., Song X., Citron M., Suzuki N., Bird T.D., Hardy J., Hutton M., Kukull W., Larson E., Levy-Lahad E., Viitanen M., Peskind E., Poorkaj P., Schellenberg G., Tanzi R., Wasco W., Lannfelt L., Selkoe D., and Younkin S., 1996, Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease, Nat Med 2: 864-870.
http://dx.doi.org/10.1038/nm0896-864
Suzuki N., Cheung T.T., Cai X.D., Odaka A., Otvos L., Eckman C., Golde T.E., and Younkin S.G., 1994, An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants, Science, 264: 1336-1340.
http://dx.doi.org/10.1126/science.8191290
Tomiyama T., Nagata T., Shimada H., Teraoka R., Fukushima A., Kanemitsu H., Takuma H., Kuwano R., Imagawa M., Ataka S., Wada Y., Yoshioka E., Nishizaki T., Watanabe Y., and Mori H., 2008, A new amyloid beta variant favoring oligomerization in Alzheimer's-type dementia, Annals of neurology, 63: 377-387.
http://dx.doi.org/10.1002/ana.21321
Theuns J., and Van Broeckhoven C., 2000, Transcriptional regulation of Alzheimer's disease genes: implications for susceptibility, Hum Mol Genet, 9: 2383–2394.
http://dx.doi.org/10.1093/hmg/9.16.2383
Taddei K., Clarnette R., Gandy S.E., Martins R.N., 1997, Increased plasma apolipoprotein E (apoE) levels in Alzheimer's disease, Neurosci Lett, 223: 29–32.
http://dx.doi.org/10.1016/S0304-3940(97)13394-8
Zheng H., and Koo E.H., 2011, Biology and pathophysiology of the amyloid precursor protein, Mol Neurodegener, 6: 27.
http://dx.doi.org/10.1186/1750-1326-6-27
Zhou L., Brouwers N., Benilova I., Vandersteen A., Mercken M., Van Laere K., Van Damme P., Demedts D., Van Leuven F., Sleegers K., Broersen K., Van Broeckhoven C., Vandenberghe R., and De Strooper B., 2011, Amyloid precursor protein mutation E682K at the alternative beta-secretase cleavage beta'-site increases Abeta generation, EMBO molecular medicine, 3: 291-302.
http://dx.doi.org/10.1002/emmm.201100138
Zhong N., and Weisgraber K.H., 2009, Understanding the association of apolipoprotein E4 with Alzheimer disease: clues from its structure, J Biol Chem, 284: 6027–6031.



. PDF(427KB)
. FPDF(win)
. HTML
. Online fPDF
Associated material
. Readers' comments
Other articles by authors
. S. Pooja
. Anitha P. Muttagi
. Seema J. Patel
. H. Gurumurthy
. Prashantha Nagaraja
Related articles
. Alzheimer’s disease
. Transcriptional analysis
. Microarray
. Biostatistics
Tools
. Email to a friend
. Post a comment